A Year of COVID-19
From SARS-CoV-2 to COVID-19 Pathogenesis, Vaccines and Therapeutics
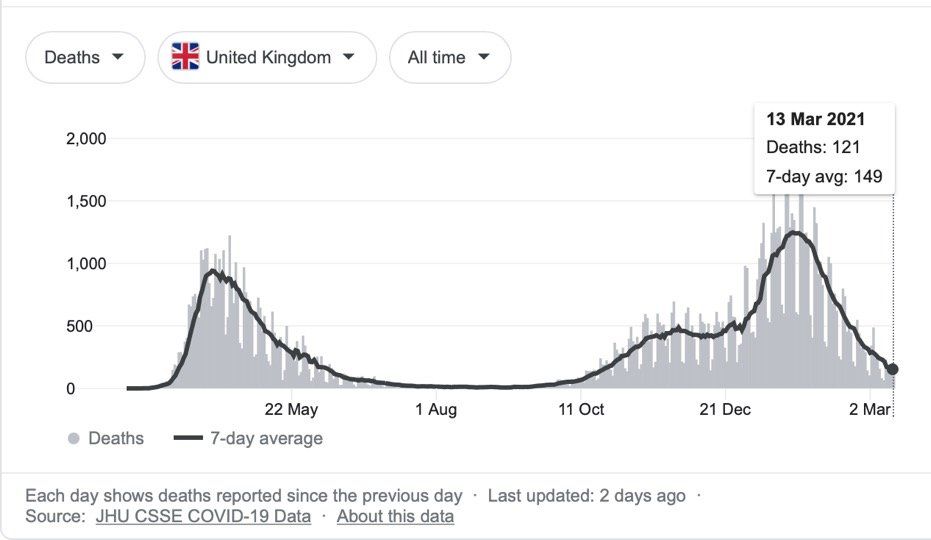
Sources: JHU CSSE COVID-19 Data and Google
On 11th March 2020, the World Health Organisation declared pandemic status for COVID-19, an infectious disease caused by the new coronavirus SARS-CoV-2. This virus was first isolated from airway epithelial cells of patients presenting with pneumonia of unknown cause in Wuhan, China, in December 2019. Since then, there has been a concerted global effort to characterise the virus and understand the pathophysiology of COVID-19, develop vaccines and search for drug therapies. As of 10th March 2021, 111,300 COVID-19 or SARS- CoV-2 citations can be found in PubMed. That number is growing daily.
A year on, the aim of this article is to give an overview of what has been learned about the SARS-CoV-2 virus (Fig. 1.) and the symptomatology and pathogenesis of COVID-19 based on a review of published literature, particularly how it relates to disease severity. Specific details may change as future research expands and refines current knowledge, but hopefully this article will remain substantially accurate for the most part.
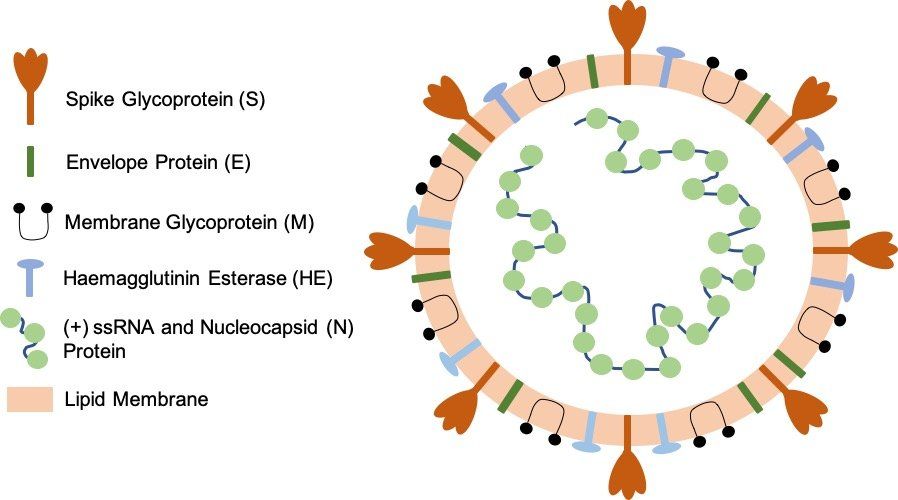
Fig. 1. SARS-CoV-2 virion particle. The coronavirus virion consists of the following structural proteins: spike glycoprotein (S), envelope protein (E), membrane glycoprotein (M), nucleocapsid protein (N) and haemagglutinin-esterase (HE). The positive-sense, single-stranded RNA genome (+ssRNA) is encapsidated by N, whereas M and E ensure its incorporation into the viral particle during the assembly process. S trimers protrude from the host-derived viral envelope and provide specificity for the cellular entry receptors ACE2 and TMPRSS2 (Fig. 2.).
TRANSMISSION AND INFECTION
Transmission and Detection
SARS-CoV-2 is spread predominantly through aerosols and airborne droplets produced by infected individuals coughing and sneezing. COVID-19 symptoms appear after an incubation period of 5 to 6 days, with peak symptoms occurring 2 to 5 days later. Virus can be detected during the incubation period by analysis of nasal or throat swab samples collected from infected individuals. A wide range of detection methods have now been developed to detect the presence of virus, with RT-PCR being the most widely used. Shedding of virus particles by infected individuals, particularly superspreaders, before symptom onset, contributes to the difficulty of controlling virus spread by public health interventions.
Infection
Infection leading to COVID-19 occurs when SARS-CoV-2 spike protein attaches to ACE2 (Fig. 2.), a cell surface protease expressed at variable levels on the surface of epithelial cells in the mouth, nose, and upper and lower airways of the lungs. After attachment to ACE2, the transmembrane protease TMPRSS2 cleaves and primes the bound spike protein to facilitate cell entry by endocytosis. Once inside the cell, the virus hijacks the host cellular machinery to replicate itself and assemble more virions. Virus particles are unpackaged and genes encoded by the viral RNA genome are translated into viral proteins by host ribosomes in the endoplasmic reticulum. Viral progeny are then assembled from the synthesised structural proteins and replicated viral RNA genome in the Golgi complex before being released from the host cell by exocytosis. One infected host cell can create up to a hundred new virions, accelerating the spread of the virus via infection of other cells in the respiratory tract.
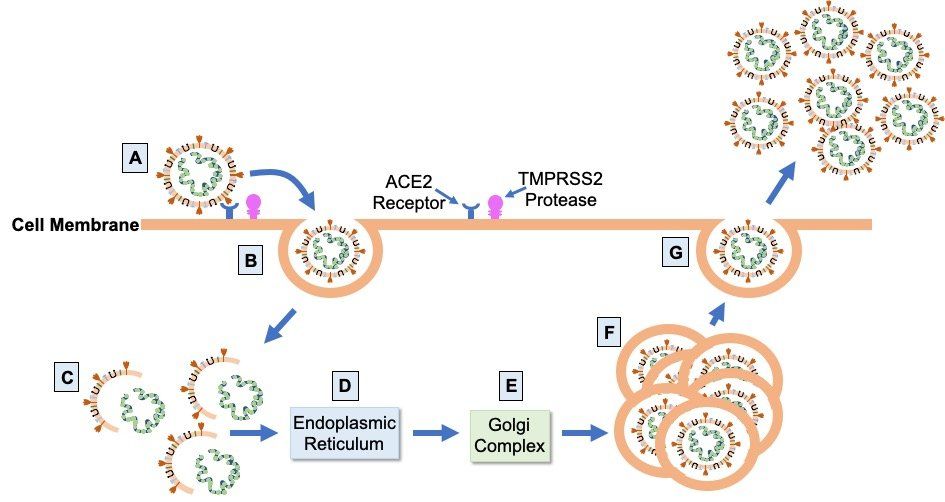
Fig. 2. SARS-CoV-2 infection cycle. Virions bind to ACE2 on the apical surface of epithelial cells in the respiratory tract via the virus S glycoprotein (A). Cleavage and priming of the bound S protein by TMPRSS2 enables endocytosis of virion particles (B). After cell entry, uncoating of the virions releases the viral genomic RNA (C) which is then translated on the ribosomes of the endoplasmic reticulum (D) to form the proteins of the viral replication and transcription complex, as well as the viral structural proteins. Translated structural proteins transit through the Golgi complex (E), where interaction with N-encapsidated, newly produced genomic RNA results in the formation of mature virions (F) ready for secretion from infected cells by exocytosis (G).
COVID-19 and the Respiratory Tract
The respiratory tract is the first line of defence against inhaled virus particles. SARS-CoV-2 shows a gradient of infectivity from the upper to the lower airways, matching the distribution of ACE2 expression levels. Cytokines released by infected airway epithelial cells and resident, plus incoming, immune cells mediate the cell-cell communication required for a tightly regulated immune response. In COVID-19, the host immune response is a critical component of disease progression and severity. Dysregulation of this response is a key feature in individuals with severe COVID-19.
COVID-19 SYMPTOMATOLOGY AND SEVERITY
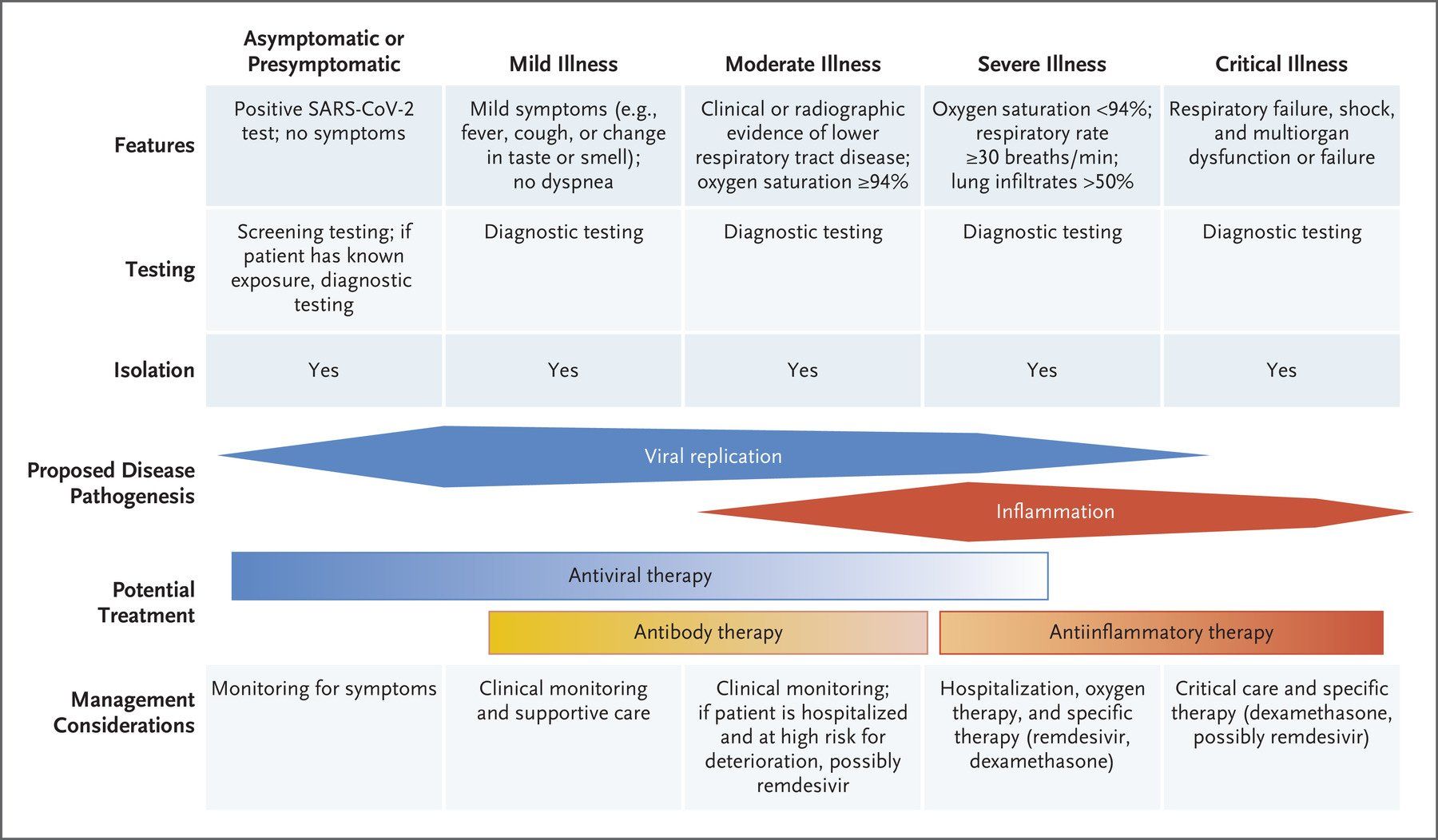
The symptoms (Fig. 3.) displayed by people infected with SARS-CoV-2 vary in type, number and severity, as well as duration (Fig. 4.). These range from asymptomatic carriage, to mild/moderate disease, to severe disease, characterised by atypical pneumonia, respiratory failure and acute respiratory distress syndrome (ARDS). The most severely affected patients tend to be older men, people from ethnic minorities and people with co-morbidities, such as obesity, diabetes and hypertension. In addition, factors such as genetics, viral load, the level of immune response and degree of lung damage will also play a part, with a predominance of different factors in different patients causing different individual clinical outcomes.
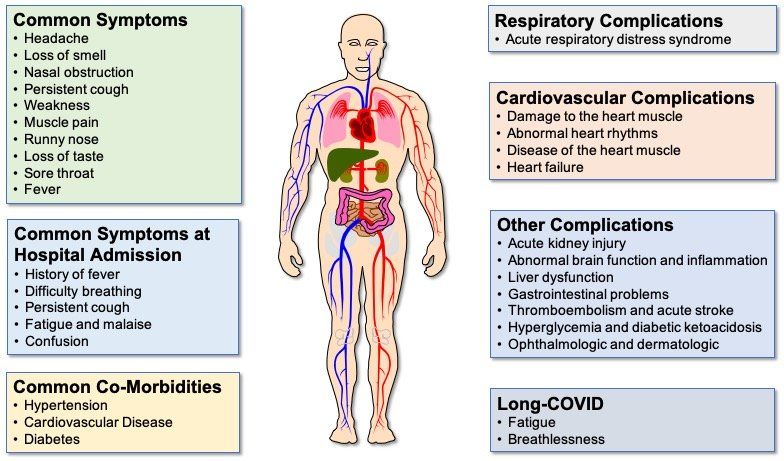
Fig. 3. COVID-19 symptoms and complications. Although fever, persistent cough and loss of taste and/or smell are the most common COVID-19 symptoms reported, some patients display a number of different symptoms. Difficulty breathing is a predominant feature of the majority of COVID-19 patients who are hospitalised. Common co-morbidities are heart disease, high blood pressure and diabetes. Although the lung is the primary site of COVID-19 pathology, complications affecting a range of extra-pulmonary organs are often seen in patients with severe disease, particularly those with co-morbidities causing endothelial cell dysfunction. Even in patients with mild/moderate disease, some patients experience long COVID, where symptoms can persist for 12 weeks or longer.
Asymptomatic COVID-19
It has been estimated that 17% of people infected with SARS-CoV-2 never develop any symptoms of COVID-19. These are not pre-symptomatic individuals who have no initial symptoms, but then go on to develop mild to moderate symptoms. These are individuals who never develop any symptoms at any time. It is not known which demographic, clinical, or immunological characteristics are responsible for the lack ofsymptomatology in these people compared with those who develop symptoms. Neither is it known the degree to which asymptomatic individuals contribute to virus transmission.
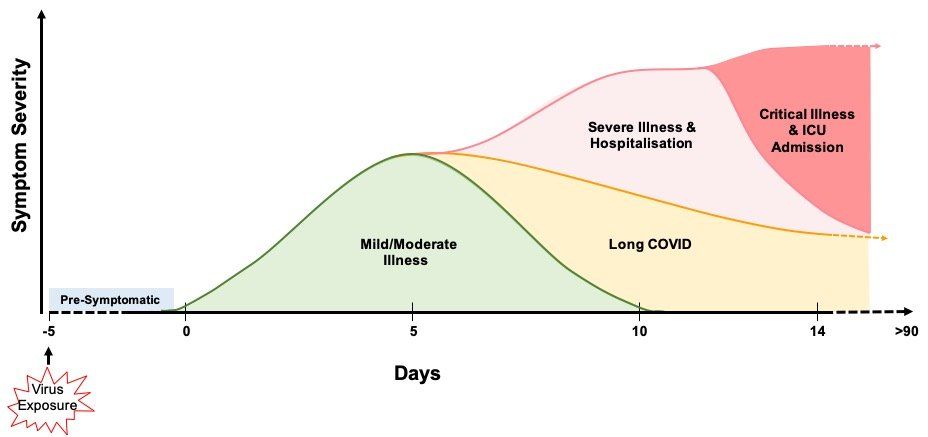
Fig. 4. COVID-19 symptomatology time course and outcomes. COVID-19 symptoms become apparent around 5 days after initial virus exposure. Symptoms peak, on average, 5 days later and, for the majority (>60%) of people, COVID-19 symptoms are mild to moderate in severity. Although symptoms subside for 4 in 5 of these mild/moderate cases after another 5 days, 1 in 5 people can experience persistent symptoms for 12 weeks or more (long COVID). Twenty percent of infected individuals require hospitalisation with breathing difficulties caused by viral pneumonia. Of these hospitalised patients, 1 in 5 develop ARDS and/or multi-organ failure requiring intensive care. Up to 40% of critical care patients can die. It has been estimated that around 17% of infected people never develop any symptoms of COVID-19 at any time.
Mild to Moderate COVID-19
The majority (> 60%) of infected individuals develop mild to moderate disease symptoms that generally resolve spontaneously after 6-10 days (Fig. 4.). However, around one in five of these individuals can have symptoms that persist from 4 weeks to over 12 weeks. Whether this post-COVID syndrome, generally known as long COVID, is a result of: long-term persistence of the virus; long term sequelae resulting from disruption of individuals’ immune and inflammatory responses; or due to something else entirely, is currently unknown.
Severe COVID-19
Around 20% of infected individuals require hospitalisation (Fig. 4.). Viral pneumonia, as evidenced by abnormal temporal lung changes on chest radiography, is the most common reason for admission, with patients displaying hypoxia due to a drop in blood oxygen levels. This leads to the majority of admitted patients suffering respiratory failure and requiring breathing support, which varies depending on individual patient need. Eighty percent of hospitalised patients are cared for in general medical wards.
Critical COVID-19
Twenty percent of hospitalised patients become critically ill and require invasive lung ventilation and support in high dependency and intensive care units (Fig. 4.). Sixty to eighty percent of these patients have ARDS and a hyper-inflammatory response. Unfortunately, up to 40% of critically ill COVID-19 patients die, although the death rate does vary with age. Severe COVID-19 can result in permanent damage and scarring to the lungs in survivors.
Multi-Organ COVID-19
Although the lungs are the primary site of COVID-19 pathology, COVID-19 is a multi-system disease exhibiting a range of complications resulting from damage to a number of different organ systems, including the heart, brain, kidney, liver and vasculature (Fig. 3.). Elevations in levels of cardiac injury biomarkers are observed in 50% of patients with severe COVID-19, with a significant proportion of patients developing venous and arterial complications. Individuals with pre-existing endothelial cell dysfunction have an increased risk of damage caused by SARS-CoV-2 infection, which also increases hypercoagulability and results in a high incidence of venous thromboembolism and pulmonary embolism. Less commonly, SARS-CoV-2 infection can cause heart muscle inflammation and heart rhythm disturbances, such as atrial fibrillation. In cases of multi-organ dysfunction, specialist clinical support to reduce multi-organ failure and mortality is required.
COVID-19 PATHOGENESIS
The upper airways
The nasal cavity is the first site of infection (Fig. 5A.). Here, SARS-CoV-2 infects ciliated and secretory cells which express ACE2 and TMPRSS2. The viral load in the nasopharynx and oropharynx appears to be similar between infected people who stay asymptomatic and those who become symptomatic. There is little data at the current time relating to the extent to which SARS-CoV-2 spreads further in to the airways of asymptomatic people, but what data there is suggests the virus can spread deeper into the airways in some of these individuals and it is the host response which is a key determinant of outcome, including pre-existing immunity.
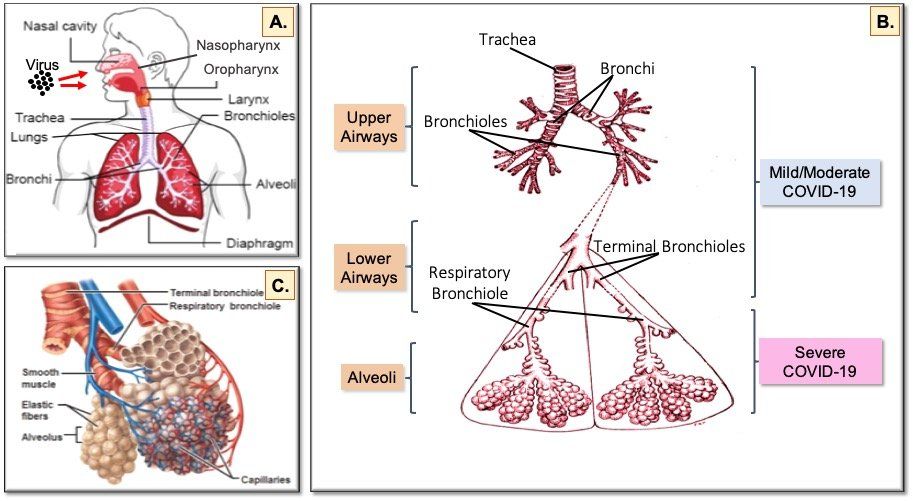
Fig. 5. The human respiratory tract and COVID-19. Inhaled virus particles bind to ACE2 receptors on the surface of epithelial cells in the nasopharynx and oropharynx (A). Disease severity is dependent on how deep SARS-CoV-2 virions progress into the airways. Restriction of virus infection to the upper and conducting airways (B) results in mild/moderate symptoms seen in >60% of infected individuals. Progression of the virus to the alveoli (C) affects exchange of oxygen and carbon dioxide between the lungs and the blood, which leads to the breathing difficulties seen in the 20% of patients hospitalised with severe COVID-19. In the 20% of hospitalised patients who become critically ill and require intensive care, damage to the endothelial cells of the capillary net encasing each alveolus leads to acute respiratory distress syndrome (ARDS) and/or multi-organ complications resulting from systemic effects of the virus.
Virus spreads from the nasal cavity to infect ciliated cells and non-ciliated mucus secreting (goblet) cells lining the bronchi (Fig. 5B.) as the virus progresses deeper into the lungs. During this phase, a vigorous innate immune response to clear the virus is triggered and the infected individual becomes symptomatic and feels unwell. Infection of lung epithelial cells triggers cell death and inflammatory responses. Keratin 5-expressing basal cells, which are the progenitor cells for the airway epithelium and are not infected by SARS-CoV-2, are induced to differentiate directly into ciliated cells so damaged epithelium can be repaired and recover from the infection. If the capacity for basal cell differentiation is impaired (e.g. due to excessive interferon lambda production), lung epithelial cell regeneration does not take place and airway damage increases, as happens in the lungs of patients with severe COVID-19.
The Lower Airways
Infection of club (Clara) cells located in the respiratory bronchioles in the lower airways facilitates spread of SARS-CoV-2 into the alveoli (Fig. 5C.), where oxygen and carbon dioxide exchange between the lungs and the blood is impaired in severe COVID-19.
Two types of epithelial cells line the alveoli (Fig. 6A.). Type I pneumocytes are large flat cells that are directly adjacent to the endothelial cells of the capillary network that closely encircles each alveolus (Fig. 5C.). Type I pneumocytes are critical for rapid exchange of oxygen and carbon dioxide between the blood and alveolar space. These cells are easily damaged in many forms of lung injury. Type II pneumocytes make and secrete pulmonary surfactant, which is required for effective gas exchange. Type II pneumocytes are also the progenitors of type I pneumocytes and are therefore important in the repair of alveolar damage. Both type I and type II pneumocytes help keep the alveoli free of fluid by contributing to active resorption of alveolar fluid and trans-epithelial ion movement. Type I and type II pneumocytes both express ACE 2.
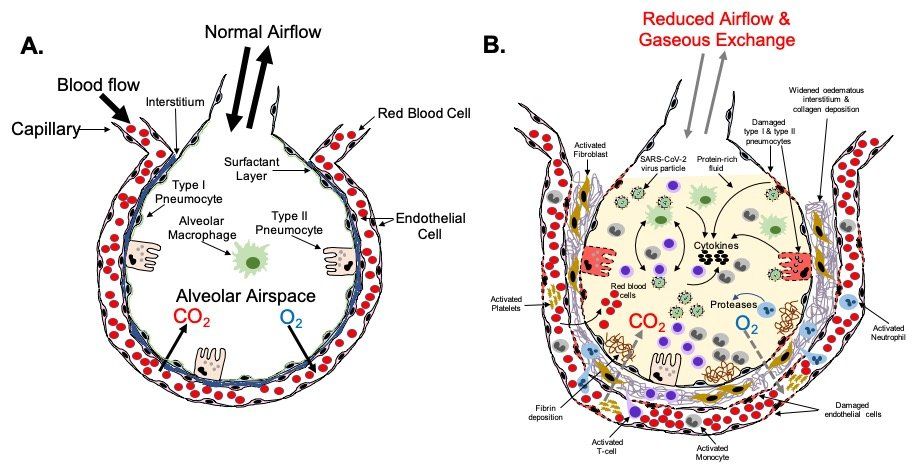
Fig.6. Severe COVID-19 lower respiratory tract pathophysiology. The alveolus (A) is the lung structure where gaseous exchange with the blood takes place. Oxygen in inhaled air and carbon dioxide in the blood diffuse out of, and in to, the alveolar space, respectively, via type I pneumocytes and the endothelial cells of the surrounding pulmonary capillary network. Surfactant secreted by type II pneumocytes aids this process by reducing surface tension at the air-liquid interface in the alveolus. Resident phagocytic alveolar macrophages provide a first line of defence against inhaled pathogens and noxious particles that make it down through the airways into the alveoli. In a healthy, non-diseased lung type I and type II pneumocytes keep the alveoli free of fluid. In severe COVID-19 (B), gaseous exchange between the alveoli and blood is severely reduced. Damage to type I and type II cells by SARS-CoV-2 infection results in the alveolar space becoming filled with liquid containing dead cells, cell debris, proteins, invading immune cells, virus particles etc. In critical disease, extensive damage to the alveoli and endothelial cells of the surrounding capillaries, plus a hyperinflammatory response leads to ARDS and respiratory failure requiring prompt intensive care. In some patients this leads to death. Fibrotic changes resulting from disruption of the normal alveolar epithelial repair process lead to permanent structural changes to the lung micro-architecture that can have future health effects in some survivors depending on the scale and severity of these changes.
In addition to type I and II pneumocytes, there is a third cell type in the alveoli - alveolar macrophages (Fig. 6A.). These phagocytes are the first line of defence against infectious agents and noxious particles present in the environment that make it down through the airways to the alveoli. Like type I and type II pneumocytes, alveolar macrophages express ACE2.
Alveolar Inflammation
Type II pneumocytes express the highest levels of ACE2 and TMPRSS2 proteins and are believed to be the primary targets for SARS-CoV-2 infection in the alveoli. Type I pneumocytes and alveolar macrophages are also infected by SARS CoV-2. As the virus propagates in infected cells, released viral particles infect other type II pneumocytes and adjacent type I pneumocytes allowing spread of the virus to adjacent alveoli. Infected type II pneumocytes and alveolar macrophages secrete cytokines to recruit immune cells from the blood into the alveolar space to destroy virus-infected cells and extracellular virus. Uninfected neighbouring cells amplify this response by responding to, and secreting, additional cytokines. The innate immune response provoked in the pulmonary parenchyma is characterized by the recruitment of bone-marrow-derived monocytes to the alveolar space and their differentiation into alveolar macrophages.
The immunological hyper-response associated with COVID-19 ARDS appears to be a key driver of severity and death in patients. Although this hyper-inflammation is often referred to as a “cytokine storm” in a large number of publications, some investigators have questioned the relevance of this term in relation to COVID-19, given the cytokine levels actually measured in patients. Alveolar macrophages have a key role in initiating and propagating this hyper-inflammatory phenotype. Infected alveolar macrophages and activated T-cells recruited from the blood form a positive feedback loop that drives persistent alveolar inflammation. Neutrophils also appear to be important in the immune response and amplifying the damage seen in the lungs of patients with COVID-19 ARDS.
Alveolar Damage
Loss of type I and type II pneumocytes due to infection and subsequent cell death results in reduced gaseous exchange in diseased portions of the lungs (Fig. 6B.). Death of pneumocytes also results in focal alveolar flooding, causing the characteristic radiographic images seen in severe COVID-19. This is a result of a combination of factors including: reduced active resorption of alveolar fluid; reduced active transport of sodium from the alveolar space into the surrounding interstitium; damage to the endothelium of adjacent capillaries causing leakage of fibrinogen and other plasma proteins into the alveolar space; and formation of fibrin exudates. The flooding impairs the ability of pulmonary surfactant produced by type II pneumocytes to adsorb to the surface of the alveoli, thus increasing surface tension, and reducing oxygen exchange. As alveoli become blocked and collapse, appositional atelectasis occurs leading to loss of secondary lobules.
In most forms of lung injury, type II pneumocytes proliferate and differentiate into type I pneumocytes leading to restoration of the alveolar epithelium. However, the diffuse alveolar damage (DAD) seen in severe COVID-19 means that this repair process is disrupted because of depletion of type II cells. Activation of alternative pathways for epithelial repair in the most severe disease is likely to be the cause of the scarring and residual disease seen in some COVID-19 survivors. As type II pneumocytes inhibit fibroblast proliferation and the expression of extracellular matrix genes in fibroblasts, loss of these cells is likely to trigger fibrosis as fibroblasts migrate into the alveolar lumen. Imbalance in the renin-angiotensin system in COVID-19 may also favour lung fibrosis. The landscape and timescale of these changes will vary as new areas of the lung get infected and the innate and acquired immune systems respond.
Perivascular Damage
A key part of COVID-19 disease progression is damage to the pulmonary endothelial cells that are adjacent to the infected and damaged alveoli (Fig. 7). Analysis of lungs from patients who died of COVID-19 has highlighted several perivascular features, including: severe endothelial injury and disrupted endothelial cell membranes associated with intracellular SARS-CoV-2 virus; widespread vascular thrombosis with microangiopathy and occlusion of alveolar capillaries; and significant new vessel growth through intussusceptive angiogenesis. Changes in the pulmonary vasculature on chest computed tomography (CT) appear to be predictive of adverse clinical outcomes in COVID-19 patients.
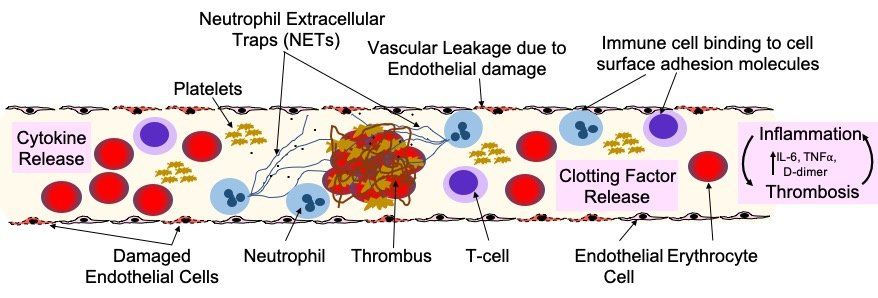
Fig. 7. Endothelial damage and dysfunction in COVID-19 lung pathology and extra-pulmonary organ damage. Endothelial dysfunction is associated with the development of severe COVID-19 and life-threatening extra-pulmonary complications. Endothelial cells can be damaged and activated by: direct viral infection; by pathogen-associated molecular patterns (PAMPs) derived from SARS-CoV-2; damage-associated molecular patterns (DAMPs) from dead or dying endothelial and immune cells; or pro-inflammatory cytokines (e.g. interleukin 6/IL-6 and tumour necrosis factor alpha/TNFa). Endothelial dysfunction and injury lead to: increased vascular permeability; increased endothelial inflammatory response; reduced levels of nitric oxide, causing vasoconstriction; and impaired angiogenesis. Activated endothelial cells release cytokines that attract immune cells that bind to adhesion molecules expressed on the surface of the endothelial cells and also produce cytokines. This stimulation of the immune response leads to hyper-inflammation, which leads to further endothelial damage. Release of clotting factors (Von Willebrand Factor, thrombin, fibrin and tissue factor), platelet activation and NETosis of neutrophils, drives thrombus formation and increases in the levels of D-dimer. Neutrophils also amplify endothelial damage by releasing a number of proteolytic enzymes and reactive oxidant species that damage cells.
Pulmonary endothelial cells (ECs) have a key role in optimising gas exchange, controlling barrier integrity and function, and regulating pulmonary vascular tone in health and disease. They are involved in most lung diseases either as a direct participant, or as a victim of collateral damage. Importantly, endothelial cells express ACE2, so SARS-CoV-2 may alter vascular homeostasis by directly infecting endothelial cells. Analysis of patient tissue samples and organoids show that SARS-CoV-2 can infect endothelial cells, even though cultured primary endothelial cells are resistant to infection.
Alternatively, instead of direct infection by SARS-CoV-2, vascular damage in COVID-19 may be driven by hypoxia, hyper-inflammation and immune dysregulation. Platelet-neutrophil communication and activation of macrophages can induce a range of pro-inflammatory effects, including: cytokine release; the formation of neutrophil extracellular traps (NETs); increasing fibrin generation; and inducing microthrombus formation in the microvasculature. NETs can damage the endothelium and activate both extrinsic and intrinsic coagulation pathways and appear to have a significant role in COVID-19 disease severity.
EXTRAPULMONARY ORGAN DAMAGE
As noted earlier, in addition to lung pathology, SARS-CoV-2 infection can result in a number of extrapulmonary complications involving the haematological, cardiovascular, renal, gastrointestinal, hepatobiliary, endocrinologic, neurologic, opthalmologic and dermatologic systems (Fig. 3.). Potential mechanisms underlying the pathophysiology of this multi-organ injury include: direct viral infection of organs; endothelial cell damage and thromboinflammation; immune response dysregulation; and dysregulation of the renin-angiotensin-aldosterone system (RAAS). The role that each of these mechanisms plays in extra-pulmonary organ damage in COVID-19 is unclear at the present time. However, the pathophysiology underlying the systemic effects of COVID-19 is likely to be multi-factorial, with different mechanisms predominating in different patients.
Direct Infection
The presence of ACE2 and TMPRSS2 in a range of different extra-pulmonary organs makes direct infection a plausible mechanism for multi-organ injury in COVID-19. Evidence for direct viral infection of the kidney and other organs in patients with COVID-19 and the detection of viral RNA in blood and urine samples from patients supports the idea of systemic spread of the virus. However, the mechanism(s) of extrapulmonary virus spread have yet to be elucidated.
Endothelial Cell Dysfunction
Endothelial cells have a number of physiological functions and are involved in a range of different diseases. Because they line the inside surface of blood vessels, they traverse a wide range of organ systems. Endothelial damage and endothelialitis are found in the vascular beds of a number of organs in patients with COVID-19 and this can trigger thromboinflammation leading to microthrombi deposition and microvascular dysfunction. Recent work suggests that inflammation and vascular damage may be the primary causes of neurological symptoms in COVID-19 patients. As a result, COVID-19 is increasingly being seen as an endothelial disease, with endothelial cell dysfunction (Fig. 7.) being the common link between many of the different complications seen in COVID-19 patients, from multi-organ damage, to blood clots and stroke. Moreover, pre-existing endothelial dysfunction is the common denominator among co-morbidities such as hypertension, diabetes, obesity, cardiovascular disease and ageing in individuals with an increased risk of severe COVID-19. Published data suggests that, at least in some COVID-19 patients, acute multi-organ thromboembolism may precede, or present disproportionately, over respiratory involvement. Organ dysfunction is also associated with excessive NET formation and vascular damage.
Immune Dysregulation
As mentioned previously, the host immune response is important in governing individual patient outcomes in COVID-19, whether that is a reduced response, or an over-enthusiastic response by the innate immune system. Markers of inflammation are predictive of critical illness and mortality in COVID-19 patients, supporting the idea that hyperinflammation is a driver of multi-organ failure in COVID-19. The positive effects of dexamethasone and IL6 receptor antagonist therapy also indicate the importance of the immune response in disease progression and severe pathology.
Renin-Angiotensin-Aldosterone System (RAAS) Dysfunction
ACE2 is a key regulator of RAAS in cardiovascular and pulmonary homeostasis. Vascular damage-induced by SARS-CoV-2, combined with pre-existing endothelial dysfunction caused by hypertension, diabetes, obesity, or age appears to be a contributory factor in COVID-19 related morbidity and mortality, including the vasculopathy and coagulopathy seen in COVID-19. Therefore, dysregulation of the RAAS offers another plausible mechanism for SARS-CoV-2 tissue damage of extra-pulmonary organs.
COVID-19 VACCINES AND THERAPEUTICS
As of 12th March 2021, there have been 118,742,439 COVID-19 cases worldwide, resulting in 2,632,955 deaths. In the UK there have been 4,241,677 cases and 125,168 deaths, although the number of cases and deaths has declined markedly from the late January 2021 peak, as a result of lockdown measures and an efficient vaccination programme.
Twelve vaccines have been approved at the time of writing. Thanks to the rapid availability of the SARS-CoV-2 genome sequence and advances in vaccine technology, the speed of COVID-19 vaccine development has been remarkable, taking less than a year from genome sequence determination to the UK approving the Pfizer-BioNtech vaccine, quickly followed by approval of the AZ-Oxford vaccine. This was a timescale previously unimaginable using traditional vaccine development methodology.
As of 10th March 2021, 23,053,716 people, representing 34.59% of the UK population have received at least one dose of the Pfizer-BioNtech or AZ-Oxford vaccine. A smaller number, 1,351,515 people, representing 2.03% of the UK population, have been fully vaccinated after receiving two doses. The number continues to grow daily.
The roll out of SARS-CoV-2 vaccines is already having a major impact on the incidence of severe COVID-19. The emergence of new SARS-CoV-2 variants that have increased rates of transmission and/or may reduce or abolish the effectiveness of the current vaccines is a concern. However, global surveillance of virus variants by sequencing, combined with the ability to quickly develop new vaccines, will reduce this threat. As the risk of new variants is lowered if the rate of new infections (and viral replication) is reduced, equitable access to vaccines for the world’s population, particularly poorer countries, will also be very important in reducing the number of new variants in the medium to long-term.
Can SARS-CoV-2 be eradicated, or will we just have to learn to live with it? I suspect the latter is the more achievable scenario, at least in the medium term, given: the level of vaccine hesitancy that exists; the proportion of people in the world who have still to be vaccinated; and the continuing emergence of virus variants.
The global research effort focussed on SARS-CoV-2 and COVID-19 has been unprecedented. In the time it has taken to write this article, over 8,000 new papers have been added to the 103,276 papers that existed in PubMed when I started writing! However, in contrast to the situation with vaccines, only modest progress has been made with regard to the development of anti-viral drugs to prevent SARS-CoV-2 infections, and other drugs to stop COVID-19 disease from worsening. This lack of success is not due to lack of effort, just a reflection of the scale of the problem that COVID-19 has presented to the scientific and medical worlds. In truth, drug discovery has still to reach the level of speed and refinement that vaccine technology currently has. So far, remdesivir is the only anti-viral drug approved for treating Covid-19, but its use is limited to clinical settings and, at best, it appears to be only modestly effective. Emergency use applications (EUAs) have also been issued by the US Federal Drugs Administration (FDA) for several unapproved monoclonal antibody therapies to treat mild to moderate COVID-19 in children and adults. In terms of immunomodulators for severe COVID-19 disease, dexamethasone and tocilizumab, particularly when used together, have been shown to reduce COVID-19 mortality.
How can the world be better prepared for similar threats in the future? Preparation for the next crisis occurs needs to begin now. Pre-emptive development of effective and readily available drugs that can be quickly tested in new diseases as they arise is required, mirroring the pre-emptive strategies that allowed the rapid development of SARS-CoV-2 vaccines. The last point notwithstanding, there will always be a lag between the outbreak of a new pandemic and the delivery of an effective vaccine, so antiviral drugs will be the primary tools that can be used to help keep populations safe during this period. There will also be a need for immunomodulatory drugs to treat severe disease in susceptible individuals and reduce mortality. In the main, I believe this can be most effectively approached using drug re-purposing strategies, rather than starting drug discovery programmes from scratch. This is an opportunity for pharmaceutical companies to work together with government agencies and academia in public-private consortia to pool resources and expertise to form a centralised library of drug compounds that can be rapidly tested in clinical trials. Furthermore, investment in identifying future primary threats and making a societal commitment to pre-emptive preparation is also needed. This will require a high degree of coordination and the involvement of scientists from fundamental researchers, to drug development experts. Given the remarkable response to the current pandemic, this is achievable. Ideally, the work needs to be undertaken globally, to spread the cost and maximise expertise and resources, as well as ensure that important research on other diseases is not disadvantaged.
What about the use of viral challenge studies in the development of new anti-viral drugs and vaccines? Approval of the world’s first challenge study in the UK is a key step towards this goal. If such studies can be conducted safely on healthy volunteers they will be important in facilitating the development of new vaccines and therapeutics. However, they will not be of value in the development of drugs to target severe disease as it would clearly be unethical to deliberately trigger life threatening symptoms in volunteers. For this, standard clinical trials using patients hospitalised with severe disease will still be required, underlining the importance of having libraries of immunomodulatory drugs ready for rapid testing in such studies when a pandemic arises.
CONCLUSION
Although the scientific and medical response to the COVID-19 pandemic has been exceptional, and the progress made in understanding the disease and developing vaccines has been astounding, the social and economic cost underlines the importance of learning from the current pandemic and being better prepared for the next one. Clearly, it is not a question of if, but when, it will occur again and how well the world will be prepared for it next time…